Single-Cell Data Formats in FASTGenomics
As a researcher in the field of single cell RNA-sequencing, new datasets are always exciting to work with. However, it can be challenging to analyze datasets from public sources as they come in a variety of formats. Sometimes, you download a set of files in an uncommon, non-standard format, or data that are only readable using a specific software package. Converting between formats is often prone to information loss or requires installation of different tools. Have you been there? We feel you. It is our vision at FASTGenomics to take care of software installation and data loading so that you can focus completely on what is important – your research.
In this article, we will explain how to use FASTGenomics’ provided readers out of the box, and how to implement your own reading routine if you are using a file format FASTGenomics does not yet support.
Which single-cell data formats are supported by FASTGenomics?
The key to data loading in FASTGenomics are our reader modules fgread-r and fgread-py. They implement reading of various standardized single-cell file formats in both R and Python. All you need to do is tell FASTGenomics the file format of your dataset and your data will be read automatically by our fgread module. Currently, FASTGenomics supports the following file formats:
Name | Extension | Description |
| 10x hdf5 | .hdf5 | 10x HDF5 Feature Barcode Matrix Format |
| Seurat object | .rds | Only supported in R, not in Python |
| AnnData object | .h5ad | For Seurat we provide a beta version since loading h5ad is not working reliable in Seurat v3 |
| Loom | .loom | For Seurat we provide a beta version since loading loom is not supported in Seurat v3 |
| tab-separated text | .tsv | Support for tab-separated, dense matrices with cell/gene identifiers in first row/column, respectively |
| comma-separated text | .csv | Support for comma-separated, dense matrices with cell/gene identifiers in first row/column, respectively |
If your dataset uses another format, please specify “Other” in the dataset detail information. Feel free to contact us or join the discussion on our Slack channel if you feel an important single-cell data format is missing!
How do I specify the file format of my dataset?
Upon uploading, FASTGenomics will ask you to specify the file format of your dataset. You can also change this information on the detail page of the dataset. This information can only be modified by the uploader of the dataset.
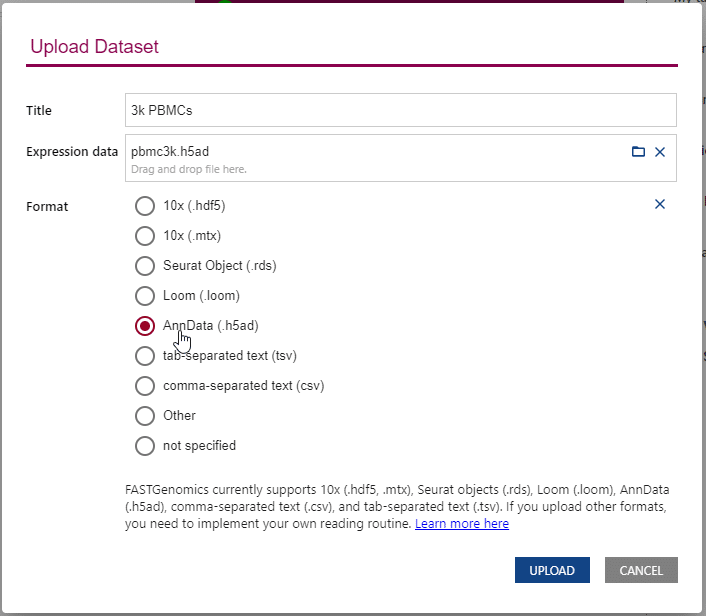
Upon uploading a new dataset, FASTGenomics will ask you to set the data format.
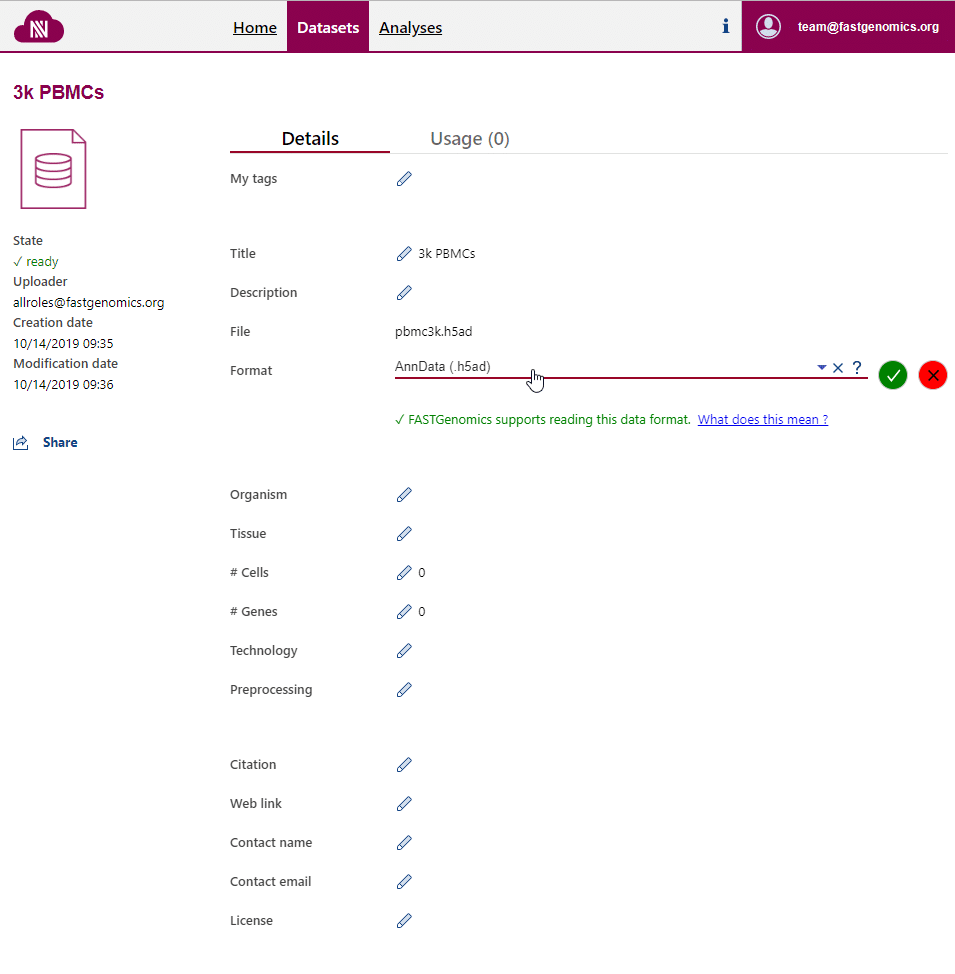
Only if the file format is specified on the dataset detail page, FASTGenomics is able to read your data out of the box.
How can I use FASTGenomics’ reader modules to load a dataset?
If your dataset uses one of FASTGenomics’ supported file formats, you can load the data using our reader modules. Simply call fgread::read_datasets() in R or fgread.read_datasets() in Python. Please note that to read AnnData and Loom in R, you need to use the option experimental_readers.
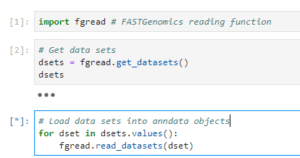
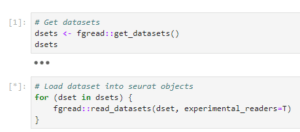
Reading datasets using fgread in R
If you are using R, the data will be loaded as a list of Seurat objects; in Python, you will get a list of AnnData objects. For details on the internal workings of our readers fgread-r and fgread-py, please visit our Github page.
Where can I get help?
If you have problems with data loading, please contact us via email or join our Slack channel. We will be happy to help!